The province of Ontario, Canada, is heading for a third wave of the Coronavirus pandemic of 2020/2021, and the reason is a yet to be identified variant of the SARS-CoV-2 virus.
As we all know by now, the Coronavirus that caused the 2020/2021 pandemic has not stayed with the same genetic sequence of the original wild type identified in Wuhan China. This is completely expected, because all living beings mutate, and viruses specially so.
Mutations: All Living Entities Evolve
All living entities are known to mutate, and by natural selection, evolve over time, to acquire new characteristics.
Any given mutation is not of significance, unless it affects the virus and/or its host (us humans in the case at hand) in one of several ways.
In fact, most mutations are neutral: they neither benefit the virus, nor harm it. Such neutral mutation are very important in the current pandemic, since it allows researchers, using genomic sequencing, to trace the different branches of the evolving variants. Public databases have been build showing which version of the virus gave rise to what version, such as NextStrain.
The mutations of significance to us humans are the ones that can:
a) increase infectiousness of the virus, causing it to spread more quickly and to many more hosts.
b) increase virulence, making it cause more severe disease for humans.
c) cause the virus to evade immunity acquired by people either naturally (prior infections) or via vaccines.
A mutation can also harm the virus, for example by interfering with how it infects hosts, such as preventing it from latching onto a given cell receptor. Or by interrupting its replication mechanism by damaging a crucial protein that is created from its genome, rendering it incapable of replication. In such cases, the mutation dies out, since it can't be propagated to another generation that goes on to replicate in other hosts. If a mutation causes the virus to be less infectious, then that variant can still propagate, until a faster spreading variant crowds it out.
For a concise explanation of variants, see Dr. Vincent Racaniello's video on virus isolates, strains and variants.
Mutation Rate Varies For Every Living Entity
The rate of mutations in viruses vary widely. The rate of mutation is measured in substitutions per nucleotide per cell infection (s/n/c). Usually, DNA viruses mutate at a slower rate than RNA viruses.
Some virus mutate incredibly fast. For example, the HIV virus that causes AIDS has the fastest mutation rate of any living entity known. That is why developing a vaccine for it is a daunting task and has not been successful so far, even after four decades of research. It is also why HIV management relies on multiple drugs taken concurrently, so as to avoid resistance to one or two of these drugs.
Influenza mutates slower than HIV, but is still very fast: that is why we need annual flu shots.
The coronavirus, as far as the rest of that virus family goes, mutates slower than influenza. One of the Four coronaviruses that cause common cold have been shown to partially evade immunity year over year, though blood plasma will still neutralize them partially, until several years have passed. We hope that SARS-CoV-2 will be like that, and we get booster shots every few years.
Variants Of Concern
Several so called Variants of Concern (VOCs) have evolved and accumulated mutations that cause these variants to be a) more infectious, b) more virulent and/or c) evade prior immunity.
The currently known variants are:
B.1.1.7: first identified in Kent, UK
Identified in Sept 2020, and found to be more contagious, as well as more virulent (makes people sicker), but no effect on immunity, so vaccines work as expected.
B.1.1351: first identified in South Africa
Less data available on it, but it has the E484K mutation which affects prior immunity, so vaccines are less effective, but still effective. Janssen and Novavax tested their vaccine in South Africa while this variant was running wild, and there is reduced efficacy, but still effective.
P.1: first identified in Brazil
Though identified in Brazil, it was first detected in Japan, in three passengers arriving there from Brazil.
Also has the E484K mutation, and hence evades immunity. It is implicated in why Brazil saw a huge second wave, despite estimates putting those who got infected in the first higher than 65% (can't remember the exact figure).
So these are the known variants, and the data is not complete on their infectiousness, virulence and immune evasion.
But there are other variants that are yet to be characterized and researched. For example, there is the recently identified B.1.526 variant first identified in New York
Ontario Variant: To Be Identified
And there is a variant in Ontario, Canada that is not yet identified, and is spreading very fast, crowding out all other variants.
What is this claim based on? From provincial public health data:
See the graph in the daily report, figures 5 and 6.
More specifically:
Here is figure 5 from the above report, showing that daily infections from variants (blue) is growing, while infections from the original strain (green) is constant:
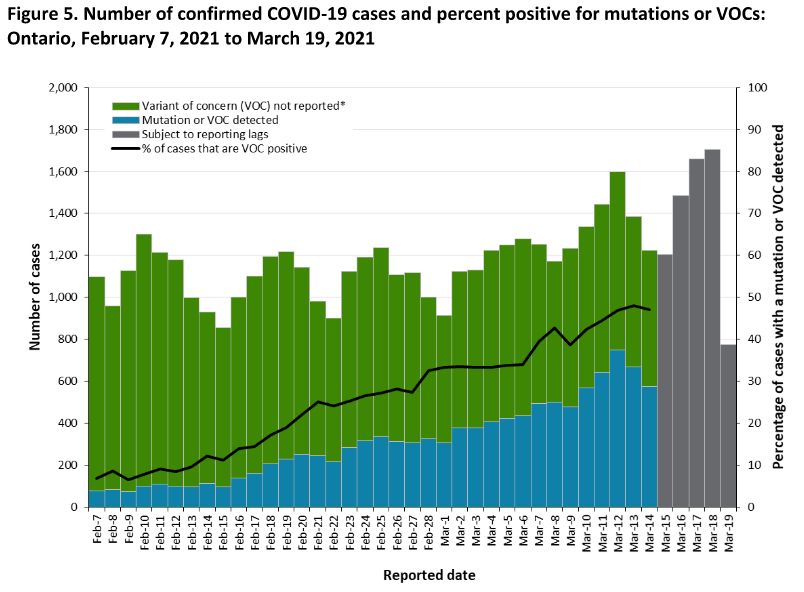
And here is figure 6, which shows that of all the positive samples that have a mutation, the vast majority, as well as the fast growing one, is not from the known three variants above (neither B.1.1.7, B.1.1351, nor P.1).
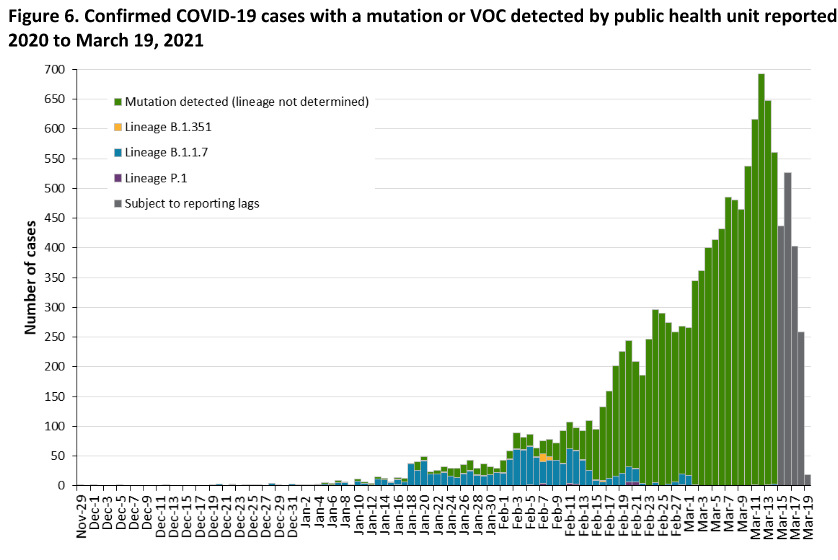
It may turn out to be the B.1.526 variant, or something new.
What is definitive is that it is causing a third wave, and we are at the beginning of it.
A Third Wave Is Not Inevitable
Although we are seeing the start of a third wave, this is not a done deal. We can avoid it if the appropriate public health measures are taken swiftly and decisively, no matter how painful these measures are.
But given the provincial government's past performance, I am not hopeful that this will ever happen.
Possible Extrapolation
What follows here is my informed opinion based on the above data.
I expect that there are much more variants out there, specially in places that have the pandemic raging on for multiple reasons. These reasons may be apathy or misinformation in the general population, distrust in health officials, lack of a social safety net for those who rely on day to day work for sustenance, and COVID fatigue ...
These variants are out there, but simply not detected, partially because not much genomic sequencing is being done. The UK sequences 10% of all positive samples, and that is why they detected the B.1.1.7 early on: because of superior genomic sequencing. But places like South Africa, Brazil (note that their variant was discovered in Japan), and perhaps India and some African countries must have variants all over the place.
Past experience shows that ignoring places with high incidence rate of a disease is problematic for the whole world, not just that country or jurisdiction. Case in point: the AIDS epidemic was left to fester in South Africa for various reasons, and the strains that evolved there went on to other parts of the world.
We are just seeing evolution happen before our eyes, and most of us just participate gladly in it by refusing to wear masks, not avoiding contact with others, believing virus, disease and vaccine misinformation, and general apathy towards the virus and pandemic.
Update 2021-03-24: What I predicted above has happened, and India has now detected a variant with two mutations: E484Q and L452R. Note that one of them is in the same position of E484, but instead of amino acid E at position 484, it is replaced with Q instead of the usual K mutation that was detected in both South Africa and Brazil, and implicated in immune system evasion.
Update 2021-03-24: According to the CBC, the growth in the unknown mutation is due to reporting lags, when samples are waiting for full genomic sequencing, and therefore is not really the result of an unknown variant. On Monday 22nd of March, the province changed the screening process to speed reporting: Samples found to have the mutation common to all three variants (N501Y) -- but not a second mutation that's common to just the B.1.351 and P.1 variants -- will no longer require the full genetic analysis. They will simply be presumed to be a variant of concern, most likely B.1.1.7. Although this is a sound strategy, I still hope that a daily sample still gets the full genomic sequencing, so that genomic surveillance is ongoing, and detects if another variant shows up (either home grown, B.1.526, the 'double mutant' from Maharashtra, India, or something else).
Most Comments
Most commented on articles ...